Interactive bioinformatics tutorials
Learn bioinformatics from your browser. Everything runs in a sandbox, so you can experiment all you want.
Get started Learn more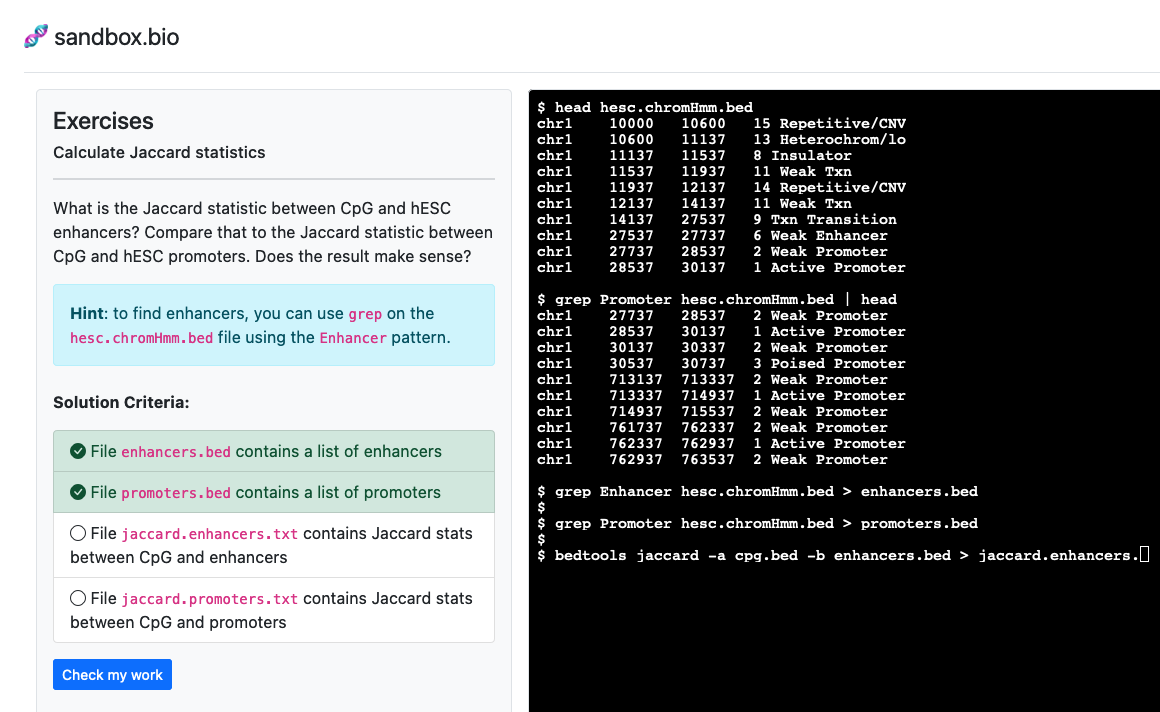
Tutorials
Recently added
Building trees with SKA
Use ska.rust to compare and align closely related small genomes using split k-mers
Wrangle FASTA and FASTQ with SeqKit
Explore and wrangle .fasta/.fastq files with SeqKit
Terminal Exercises
Command-line exercises from the Carpentries' Unix Shell lesson.
Sequence alignment with BLAST
Use BLAST to align DNA and protein sequences.
Data exploration
Terminal Basics
Get up to speed with the terminal. Start here if you're new to bioinformatics.
Terminal Exercises
Command-line exercises from the Carpentries' Unix Shell lesson.
JSON wrangling with jq
Filter and wrangle JSON files on the command-line using jq.
Data exploration with awk
Filter, extract and transform tabular data (TSV files) using awk.
File formats
Genomic intervals with bedtools
Explore and wrangle .bed files with bedtools.
BAM parsing with samtools
Explore and wrangle .sam/.bam files with samtools.
Wrangle FASTA and FASTQ with SeqKit
Explore and wrangle .fasta/.fastq files with SeqKit
Quality control
DNA sequencing QC
Evaluate the quality of a sequencing run by running fastp on your FASTQ files.
Visualize variants with IGV
Distinguish real variants from artifacts using the IGV genome browser.
Data analysis
Sequence alignment with BLAST
Use BLAST to align DNA and protein sequences.
K-mer counting with Jellyfish
Learn the basics of k-mer counting using Jellyfish
Variant calling
Use variant calling to decode a secret message stored in sequencing data.
Building trees with SKA
Use ska.rust to compare and align closely related small genomes using split k-mers
Debugging Puzzles
Debug file format issues that are commonly seen in genomics.
Playgrounds
Explore
Align DNA sequences
Explore the Smith-Waterman and Needleman-Wunsch sequence alignment algorithms.